What is CMC in Pharma?
Pharmaceutical market is highly regulated. It is usually characterized by strict approval processes and strong patent legislation.
CMC (Chemistry, manufacturing and controls) provides the foundation for a safe and effective drug. It plays an essential role in meeting regulatory requirements and ultimately driving a drug’s journey from the lab to the patient.
What does CMC mean in pharma?
The US Food and Drug Administration (FDA) defines CMC (Chemistry, Manufacturing, and Controls) as the detailed information and data related to the chemistry, manufacturing process, and quality control of a pharmaceutical product.
When pharmaceutical companies develop a new drug, a series of critical steps are necessary to evaluate and ensure its safety, effectiveness, manufacturability, and reliability. These steps, along with the associated processes, are collectively known as Chemistry, Manufacturing, and Controls (CMC). CMC encompasses the development, production, and quality assurance of the drug to ensure it meets regulatory standards and is consistently safe and effective for consumers.
The CMC Process
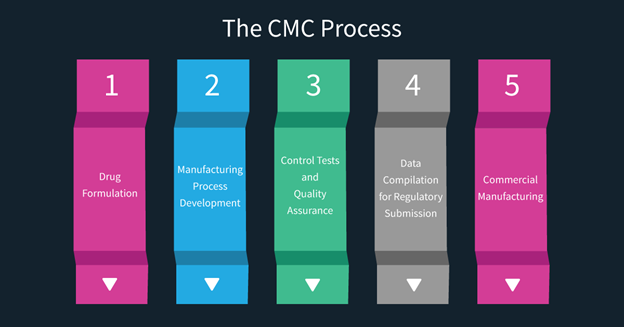
The Chemistry, Manufacturing, and Controls (CMC) process is essential to the development of a pharmaceutical product. The CMC process is divided into several key stages, from drug discovery through to commercial production. Here’s an overview of the CMC process:
1. Drug Formulation
The first step in the CMC process is drug formulation. This phase focuses on determining the composition of the drug, including both the active pharmaceutical ingredients (APIs) and the inactive ingredients. The chemistry involved ensures that the chosen ingredients are compatible and stable when combined, and that they provide the desired therapeutic effect. The formulation must be developed to maintain drug efficacy, stability, and safety across its shelf life.
2. Manufacturing Process Development
Once the formulation is finalized, attention shifts to the manufacturing process. This stage is about more than simply scaling up production—it’s about developing reproducible and efficient manufacturing methods. A consistent manufacturing process is essential to ensure that each batch of the drug meets quality standards. The manufacturing facility, equipment, and personnel must adhere to strict regulatory guidelines. Furthermore, process validation is performed to confirm that the production process yields consistent, high-quality products every time.
3. Control Tests and Quality Assurance
As the drug transitions from development to large-scale production, control strategies play a crucial role in maintaining product consistency. Various quality control tests are conducted to ensure that each batch of medicine meets purity, potency, and safety standards. These tests are designed to detect any deviations in the product’s quality.
Additionally, stability testing is a key element of this phase. It assesses how the drug holds up under various environmental conditions, such as temperature, humidity, and light exposure. Stability testing confirms the drug’s shelf life, ensuring it remains effective and safe throughout its storage period.
4. Data Compilation for Regulatory Submission
The final stage of the CMC process involves collecting and organizing all necessary data for regulatory submissions. This includes detailed documentation about the drug substance, its formulation, the manufacturing process, control strategies, and stability testing results. All of this data is crucial for demonstrating that the drug meets the required standards for safety, efficacy, and quality.
Regulatory authorities, such as the FDA or EMA, rely on this comprehensive CMC data to evaluate the drug and grant approval for market release.
5. Commercial Manufacturing
Once the drug is approved, pharmaceutical companies move into large-scale commercial manufacturing. This phase must maintain consistency in quality, potency, and safety, while meeting market demand. Ongoing quality control measures and regular audits are essential to ensure that the product remains safe and effective over time.
What are the duties of the CMC?
To give you a clearer picture of the crucial role CMC professionals play in the pharmaceutical industry, here are some key responsibilities they handle to ensure drug safety, quality, and regulatory compliance:
- CMC professionals collaborate with research, development, regulatory affairs, and quality assurance teams to create effective drug development strategies.
- A key responsibility of CMC is designing manufacturing processes that are efficient and comply with strict pharmaceutical regulations.
- CMC generates detailed documentation covering the drug’s chemistry, manufacturing methods, and quality control processes, which is crucial for regulatory approval.
- This documentation proves that the drug meets high standards for safety, efficacy, and quality, necessary for regulatory clearance.
- CMC professionals must stay updated with changing regulations and industry practices to ensure compliance and product quality.
- By maintaining high standards in drug production and quality control, CMC professionals enhance the pharmaceutical company’s reputation, earning trust from healthcare professionals and patients.
- CMC professionals ensure drugs undergo rigorous scrutiny, meeting safety and efficacy standards before reaching the market.
- Adhering to CMC guidelines is not just a regulatory requirement but an ethical responsibility to safeguard public health.
- CMC professionals ensure that drugs are scientifically validated, consistently produced, and safely controlled, benefiting patient well-being.
- CMC is the cornerstone of the pharmaceutical industry, ensuring drugs meet the highest standards from development to market, while protecting public health.
Recent CMC Considerations

Recent developments in CMC have brought new challenges and opportunities, especially with the rise of novel therapies and advanced technologies. Here are some key considerations shaping the field today:
1. Regulatory Approval Process
In the U.S., pharmaceutical manufacturers must first submit an Investigational New Drug (IND) application for approval to conduct clinical trials. Afterward, a Biologics License Application (BLA) or New Drug Application (NDA) is needed to gain market authorization.
Expedited regulatory pathways, such as breakthrough designation, fast track, accelerated approval, or priority review, are available for therapies addressing critical patient needs.
2. Role of Regulatory Agencies
The Center for Biologics Evaluation and Research (CBER) oversees biologics, including cell and gene therapies in the U.S. The Office of Tissue and Advanced Therapies (OTAT) specifically manages cell and gene therapies.
The European Medicines Agency (EMA) evaluates Advanced Therapy Medicinal Products (ATMPs) through a centralized approval process. The Committee for Advanced Therapies (CAT) provides expertise for assessing these products.
3. Guidance for Novel Therapies
Regulatory bodies, such as CBER and EMA, have developed specialized guidance documents for emerging therapies like gene therapies, cell-based therapies, and regenerative medicine, ensuring that these modalities meet specific regulatory standards.
4. CMC Documentation Requirements
Manufacturers must provide detailed documentation to demonstrate that the manufacturing process ensures consistency in the identity, purity, and potency of the final drug product. This documentation is crucial for obtaining regulatory approval.
For complex biologics, functional assays may be required to assess potency, especially when traditional methods are not reliable.
5. Manufacturing Process Control
Pharmaceutical manufacturers must establish and control critical quality attributes (CQAs) and process parameters to ensure consistent and reproducible production. This ensures that the drug maintains the desired quality throughout its lifecycle.
During technology transfers, ensuring comparability between batches becomes critical, particularly for personalized therapies like autologous cell therapies.
6. Challenges with New Modalities
For biologics such as protein-based therapeutics and gene therapies, extensive characterization is required. This includes information on amino acid sequences, post-translational modifications, and biological activity.
Gene therapy products, such as CAR T cells or CRISPR, require detailed information on the viral vectors used for gene delivery to meet regulatory standards.
7. Drug Delivery Systems
The FDA provides specific guidance for novel drug delivery systems, such as liposomes, nanoparticles, and co-formulations. Manufacturers must provide detailed descriptions of their components, physicochemical properties, and critical quality attributes to ensure safe and effective drug delivery.
8. Global Harmonization and Regulatory Challenges
As the pharmaceutical industry innovates with new therapeutic modalities, there is a lag in the development of regulatory guidance, creating challenges in meeting regulatory standards.
These differences in regulations between regions pose difficulties in global harmonization and may lead to slow approval processes, which can be resource-intensive for both sponsors and regulatory agencies.
Regulatory CMC Challenges
Now, let’s discuss some of the key regulatory challenges that sponsors face when navigating the CMC process for advanced therapies.
- Many regulatory guidelines, especially for emerging therapies like cell and gene therapies, are still in draft form, leading to uncertainty and evolving expectations.
- Traditional sterility testing methods may not be suitable for autologous cell therapies due to time constraints in patient treatment. Modified testing approaches are being explored to balance safety with the urgency of patient care.
- The lack of historical data and clear critical quality attributes (CQAs) for advanced therapies, such as gene therapies and CAR-T, makes it challenging to define a robust quality by design (QbD) framework for these modalities.
- Insufficient CMC data, particularly for new therapies, is one of the main reasons for non-approval of regulatory submissions. This is especially challenging when selecting reference standards for novel technologies.
- For autologous and patient-specific therapies, the inherent variability in starting materials and manufacturing processes complicates comparability studies, especially when using surrogate materials.
- The lack of clear regulatory pathways for novel drug delivery technologies creates challenges in moving from development to clinical and commercial phases, with added complexity in meeting quality standards.
Stay Ahead in Pharma Safety with Expert Training
The CMC ensures that pharmaceutical products are safe, effective, and meet all necessary standards before reaching consumers. CMC is an ongoing process that requires rigorous testing, process validation, and compliance with regulatory standards to ensure the safety and efficacy of the final product.
As the industry continues to evolve, staying informed and adaptable is key to success. With Metamorph’s expert-led training and events, such as our Statistical Methods for Process Validation Masterclass, you can equip your team for the challenges ahead. Stay tuned for more insights and join us in advancing your business toward lasting success.